文章来源:识林
近日CDE发布了《药品注册研发生产主体合规信息管理与审查指导原则(试行)征求意见稿》,在该文件中提出了要建立研发生产主体合规管理信息库的相关意见。
什么是注册研发生产主体合规信息?
主体合规信息似乎是以往很少提及的概念,但其实在2021年发布的药品注册核查检验启动工作程序(试行)中就已经有规定“启动药品注册核查考虑的风险因素包括品种因素和研发生产主体合规因素。其中,品种因素包括药物创新程度、药品类型、工艺和设施等,研发生产主体合规因素包括参与药学研制、临床试验、药理毒理学研究以及生产制造的相关单位和机构既往接受核查的情况等。”而合规因素的体现即本文件的主题“药品注册研发生产主体合规信息”。
意见稿中,合规信息是指能够直接或间接反映其在拟注册品种的研制过程中合规性的信息。包括但不限于:
(1)研发生产主体的基本信息和资质证明信息;
(2)在拟注册品种研制过程中承担的主要职责;
(3)接受国内外药品监管机构注册核查和监督检查的情况;
(4)对注册核查、监督检查发现问题的整改情况;
(5)研发生产主体的质量管理体系建设运行情况等。
从以上定义看,合规信息集中在研发生产主体既往提交的资料中与主体有关的信息以及既往接受核查、检查的情况,包括企业内的质量和注册部门所保存的历次官方检查(审计)的资料信息。
须注意的是研发生产主体并不限于注册申请人企业自己的检查信息,还包括和产品有关的其他合作单位的检查信息,即不单是GMP,还有GCP等检查。
合规信息有什么价值?
根据征求意见稿,CDE要“建立健全研发生产主体合规信息管理体系,以满足基于风险的核查启动工作需要。”因此合规信息的最直接价值是为CDE对申请人的风险评估提供切实全面的依据。
现在CDE实行基于风险启动药品注册核查的制度模式,即基于风险决定是否启动药品注册研制现场核查和生产现场核查。而对于申请人企业的风险究竟如何衡量和评估则是未有过很明确的指导的。
可供参照的是FDA用一种被称为场地选择模型(SSM)的风险评估工具来收集合规信息,以确定对生产场地的GMP检查的优先顺序。FDA要收集生产场地的以下信息:场地类型(例如生产商or仅包装商or仅质控实验室)、自上次监督检查以来的时间、FDA合规历史、国家或地区的合规历史、外国监管机构的检查历史、患者暴露、危害信号(例如现场警示报告、生物制品偏差报告,MedWatch报告、召回等)、固有产品风险(例如剂型、无菌制剂等)。基于收集到的数据,使用SSM为生产场地生成风险评分,进而确定生产场地检查的优先级。而现在CDE要建立的研发生产主体合规信息管理也是这样一种风险评估工具,但是覆盖面更广。
对药企后续工作的影响
在知晓了监管机构对于启动药品注册核查的风险关注点后,申请人企业也就有了标尺去评估自己的合规现状。但是对企业来说, 更重要的在于如何主动去做好自身的合规管理。
征求意见稿倡导“研发生产主体建立自身合规信息库,合规信息库应包含研制活动合规风险的评估情况,并根据后续发现合规问题的情况对风险数据进行更新。”这是一项很有价值的建议,源于在现实中可能会存在的两种情形
(1)对于多数企业来说,过往肯定是有保存着这些检查信息。但由于在面临监管机构检查以及完成后续整改行动时会有两个部门同时牵涉其中:即质量部门和注册部门。而这样就可能会出现同一次检查的所有信息被保存在两个部门且互相不知情。但如果企业内能建立一个统一的数据库就可以避免这种情况,做到全部合规信息的统一保存和管理。
(2)申请人企业对于自己的合规信息可以做到充分收集和保存,但对产品从研发走到临床结束的其他相关方的信息就会缺失。在本文件中是分别介绍了药学研制与生产、药物临床试验、药理毒理学研究三大部分的合规信息内容要求的。因此,与一个要注册申请的药品有关的主体不止申请人企业一家,至少就有下图所示的多个不同角色
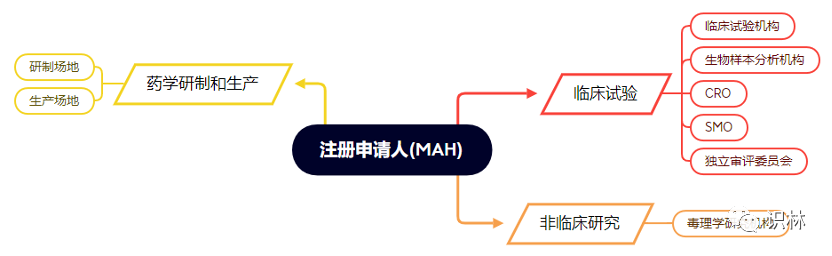
如果申请人企业内部也能有一个数据库系统,将已有的相关方的合规信息录入管理,也能起到更好的风险控制效果。不但能减少正在进行注册产品的不合规风险,也能为后续开展项目时选择合作的相关方提供更可靠的信息。
从企业实施角度的一些思考
1. 信息库建成后,与本企业产品有关的外部单位(比如临床试验机构、委托生产场地)的信息如何保持更新还未被提到。是CDE通过其他数据库获取还是外部单位自行去更新登记,还是作为药品责任主体的申请人企业进行外部单位信息的更新?如果不同企业提交了同一家外部单位的信息,但是这些信息不一致时该怎么办?
2. 征求意见稿要求的合规信息的提交是在涉及注册核查的注册申请时。这些信息未来是否也会作为其他监管行动比如临床试验申请、新药上市申请时的基础信息用以支撑申报?
3. 在药品上市后企业是否还要继续填报更新信息?合规信息如果一直在更新,是否可以扩展到影响GMP符合性检查等的风险评估?
4. 合规信息库中CRO等单位的合规信息是否会具有类似SMF(场地主文件)的作用?在信息库中已登记的企业或机构的合规信息能否被有限度的查询到,以供申请人企业在选择非临床研究、临床试验的合作单位时参考?
5. 同一企业在信息库中作为研制场地和生产场地的合规信息是否会分开登记,还是只以唯一的企业主体登记?例如某企业过去作为生产场地存在不合规信息,未来只作为研制场地时会不会还将关联到此信息而影响注册核查的风险评判?
6. 如果不同企业提交了同一家公司的信息,但是这些信息不一样该怎么办?会导致什么样的结果?
免责声明 本文系转载,仅做分享之用,不代表平台观点。图片、文章版权均属于原作者所有,如有侵权请告知,我们会及时处理。
原文链接:https://mp.weixin.qq.com/s/6R_atkFYKnI4ZTlxjKsJ5w
作者:向导@寒星苍梧


